snRNAseq annotation
For our single-nucleus RNA sequencing projects, we use an iterative clustering process to annotate the cell types. This page provides a walkthrough of our methods and a tutorial on how to perform the manual annotations.
Important
We use Seurat v4 for this tutorial.
Table of contents
Iterative clustering
The workflow begins with pre-processing the sequencing data from each sample before combining them into a single overall Seurat object. Automated annotation is first used in attempt to identify and label cell types based on reference data. The Seurat object is then iteratively integrated and clustered at the overall, class, neighborhood, and subclass levels, where manual annotation is performed at each round to try and verify the cell types.
Sample pre-processing
The samples are first processed using the Cell Ranger pipelines from 10x Genomics to generate counts data.
SoupX is then used to remove contamination from the data, and DoubletFinder is used to remove heterotypic duplicates.
Reference mapping
Reference mapping is the process of transferring cell type information from a reference dataset to our query dataset.
We use the following data sources as reference:
- Neuronal cell types from Allen Brain Atlas
- Non-neuronal cell types from Ximerakis, et al. (2019)
Integration and clustering
For most of our projects, we have data from both wildtype and transgenic mice, as well as drug treated and vehicle mice. In order to concurrently annotate data across these different experimental groups, we must first properly align these datasets in a process known as integration.
To do this, the Seurat object is split by experimental group, normalized, and recombined through integration. Specifically, we use SCTransform as the normalization method to better correct for conditional differences without removing biological effects.
Integration is performed at the overall, class, and neighborhood levels, but not the subclass level as there are often too few cells in those groups. Instead, SCTransform is performed directly on the entire subclass. Finally, the cells are re-clustered at each level and their identity is evaluated via manual annotation.
Manual annotation
While automated annotation can be a good start, it is shown that manual verification is still necessary in order to produce the best results for cell type annotations (Clarke, 2021).
The following sections provide a guide for what to look for when performing manual annotations. Generally, each cluster can be assigned to a single cell type classification; however, if there continues to be chimeric contamination at the subclass level, further subclustering may be needed in order to separate the distinct groups of cells.
Predicted cell types
A good place to start when annotating a cluster is to look at the predicted cell type of the cells that make up the cluster, as determined during the automated annotation. Note that each prediction is accompanied by a confidence score indicating how reliable it is, which can be useful to keep in mind.
Gene markers
One of the more definitive ways to verify the identity of a cluster is by looking at what genes they express. Most cell types have a unique combination of marker genes that set them apart from other types.
Use the following interactive figure to explore broad marker genes for each cell type:
We use marker genes from the following sources:
- A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation (Yao, 2021)
- Single-cell transcriptomic profiling of the aging mouse brain (Ximerakis, 2019)
- Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain (Lake, 2018)
- The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases (Krasemann, 2017)
- Disease-associated oligodendrocyte responses across neurodegenerative diseases (Pandey, 2022)
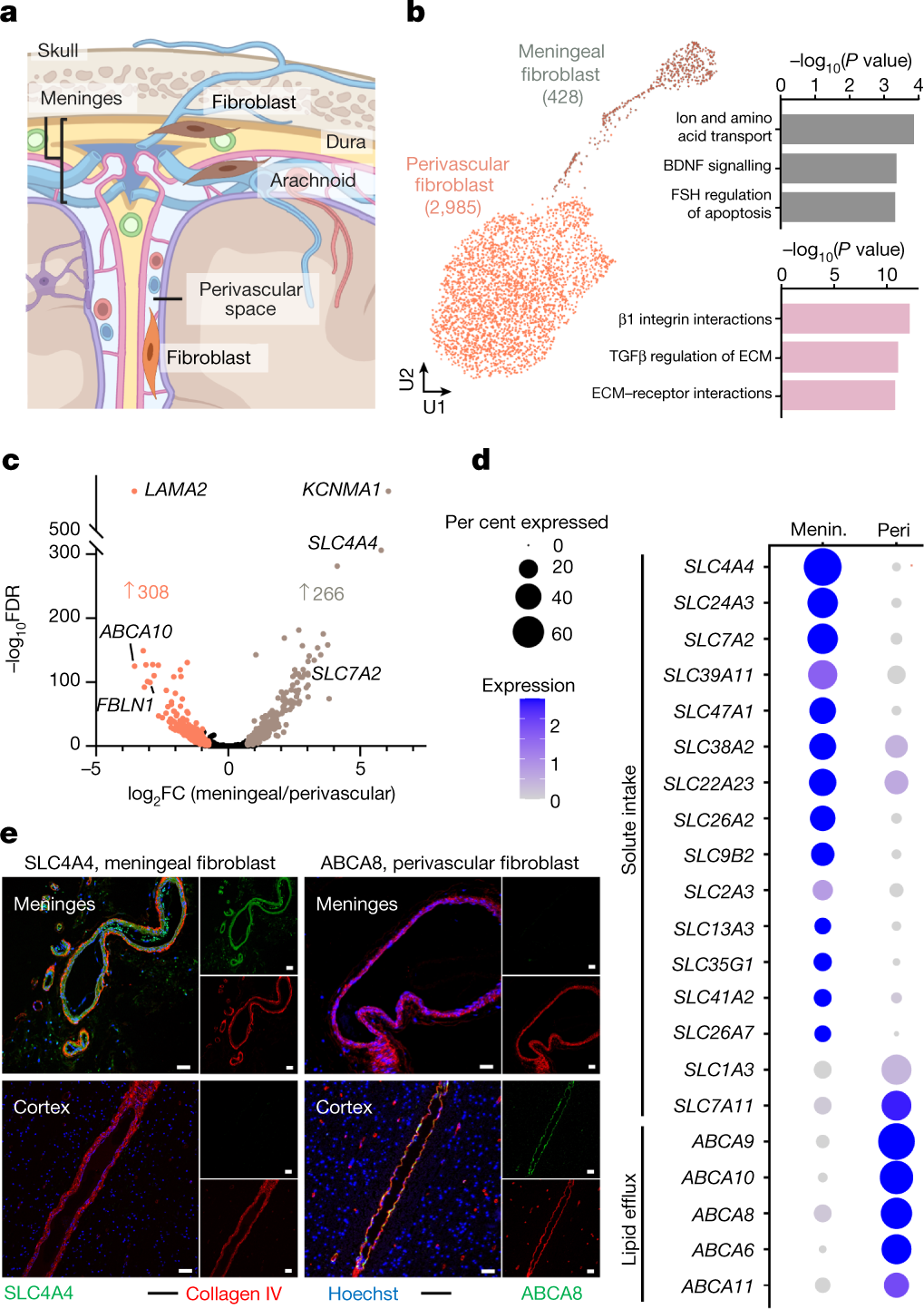
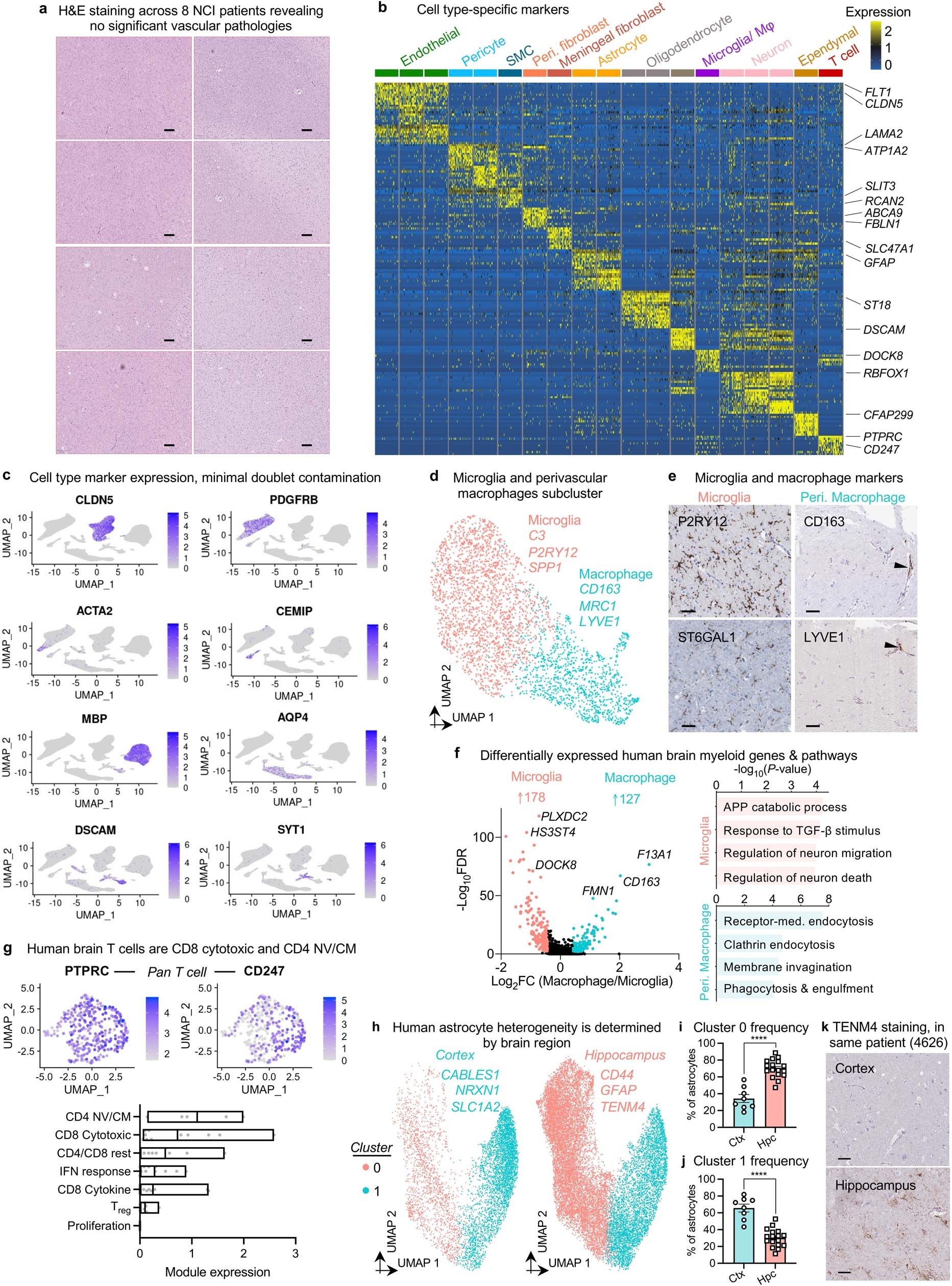
- A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk (Yang, 2022)
Note
The GABAergic class contains the CGE and MGE neighborhoods.
Caudal ganglionic eminence (CGE)
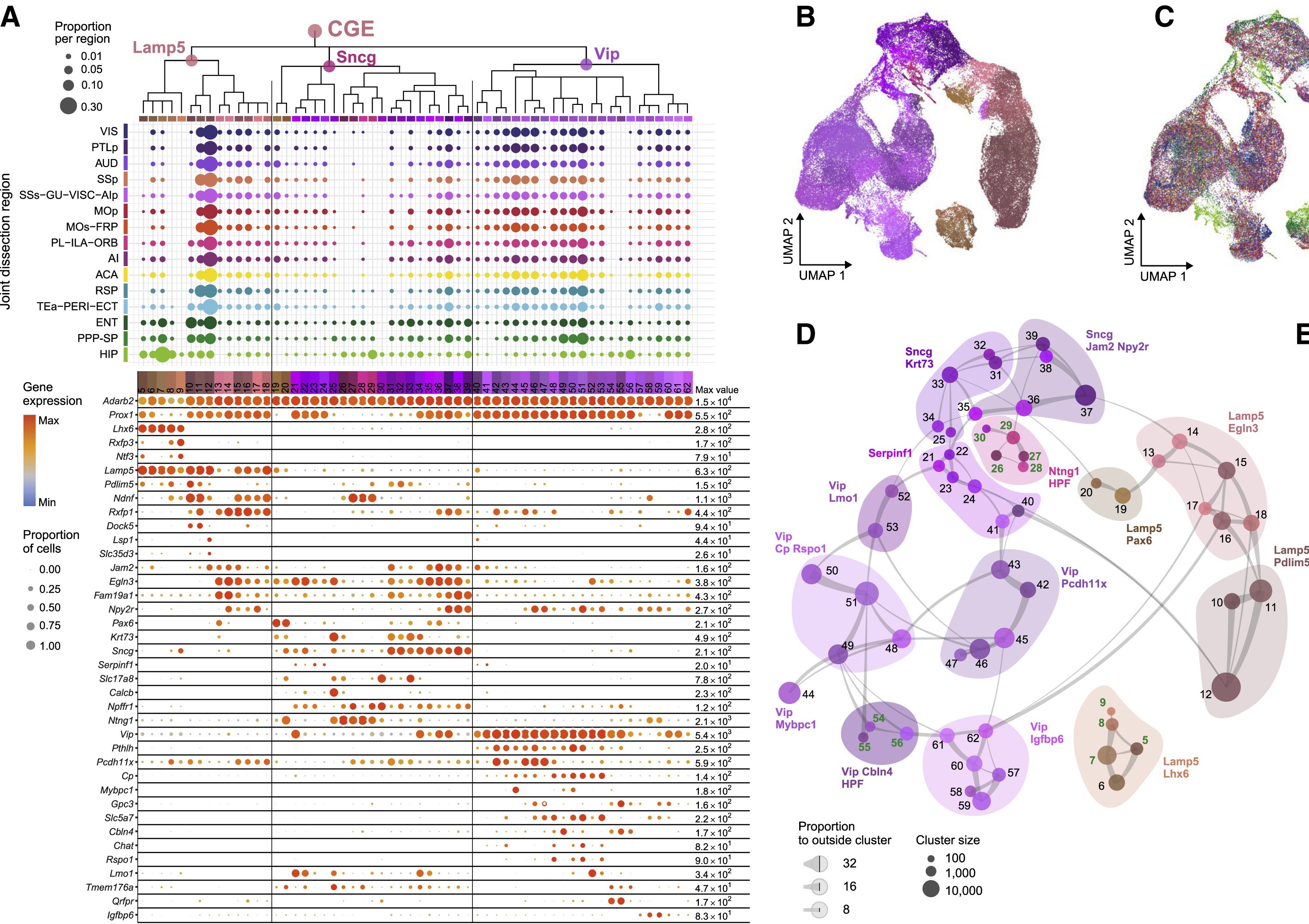
Medial ganglionic eminence (MGE)
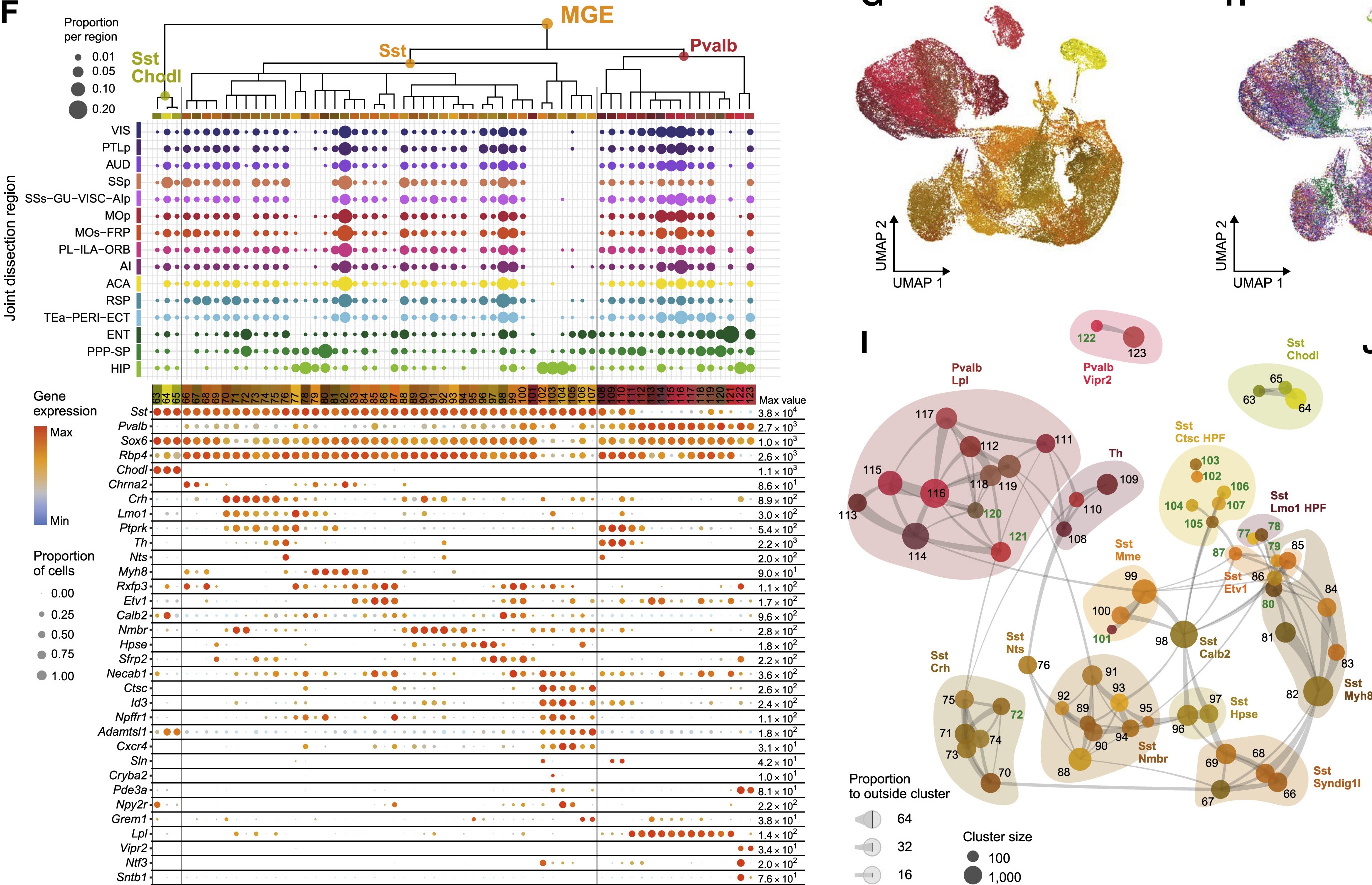
Note
The Glutamatergic class contains the L2/3 IT, L4/5/6 IT Car3, NP/CT/L6b, and PT neighborhoods.
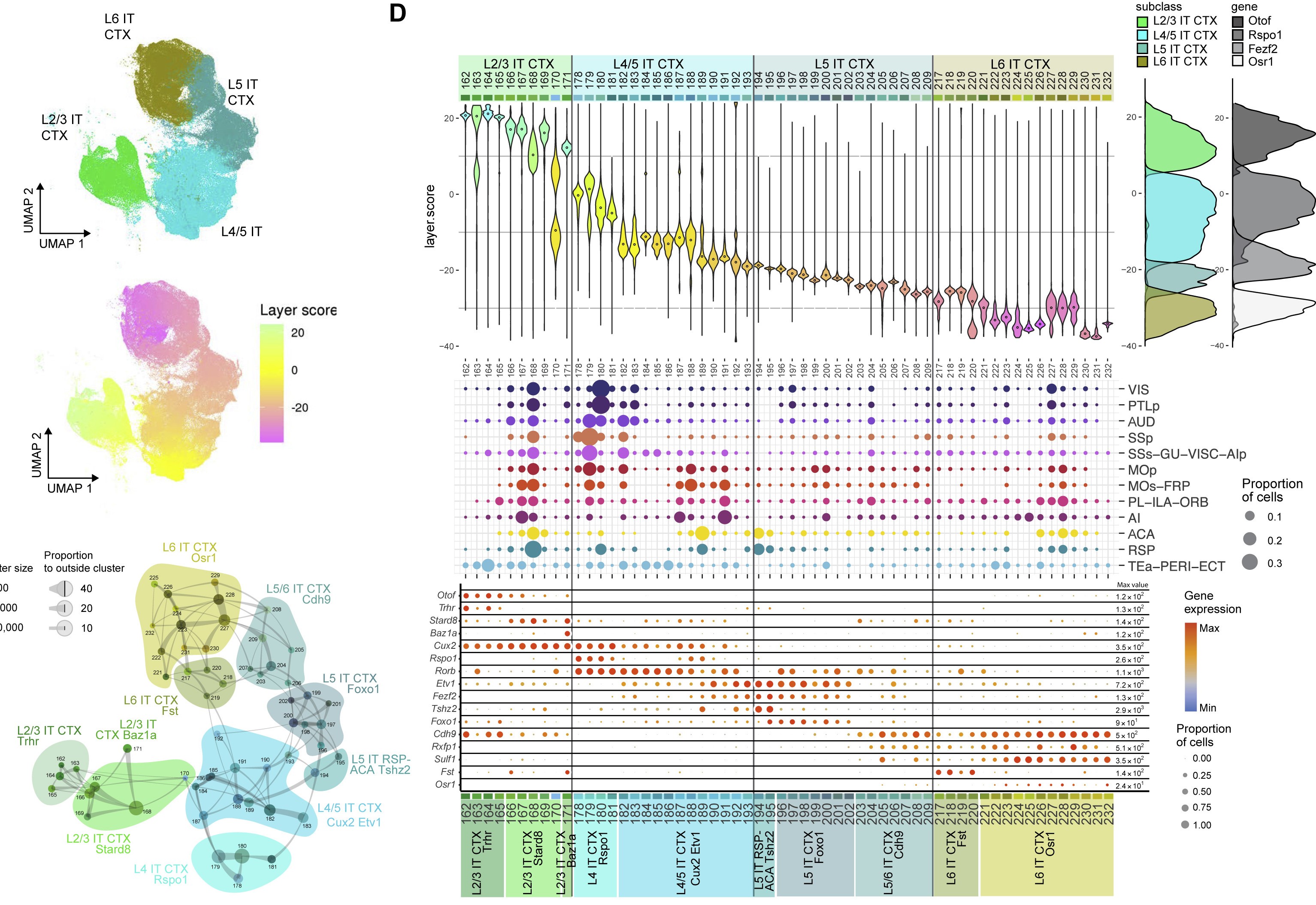
Layer 2/3 intratelencephalic neurons (L2/3 IT)
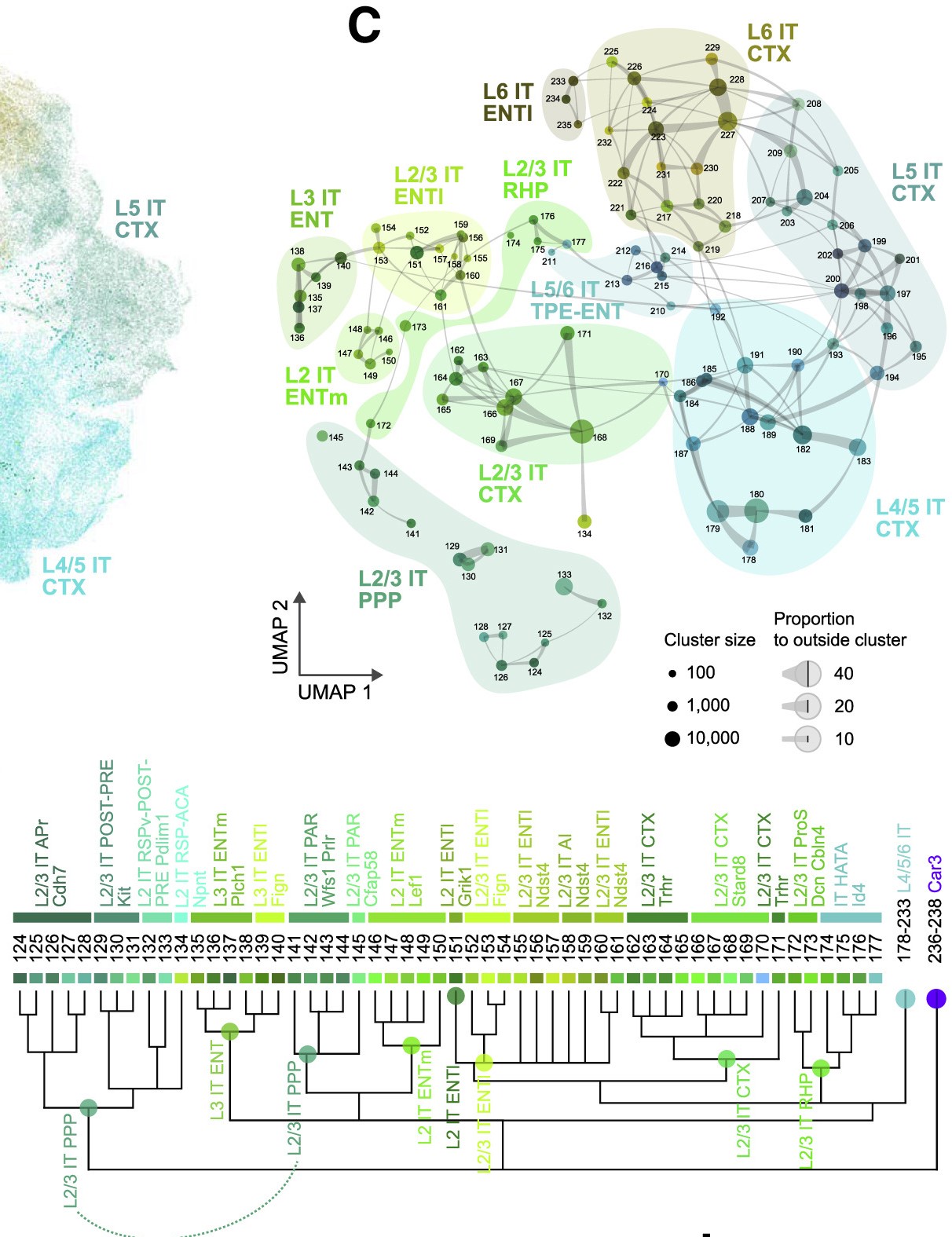
Layer 4/5/6 intratelencephalic & Car3 neurons (L4/5/6 IT Car3)
Near-projecting/Corticothalamic/Layer 6b neurons (NP/CT/L6b)
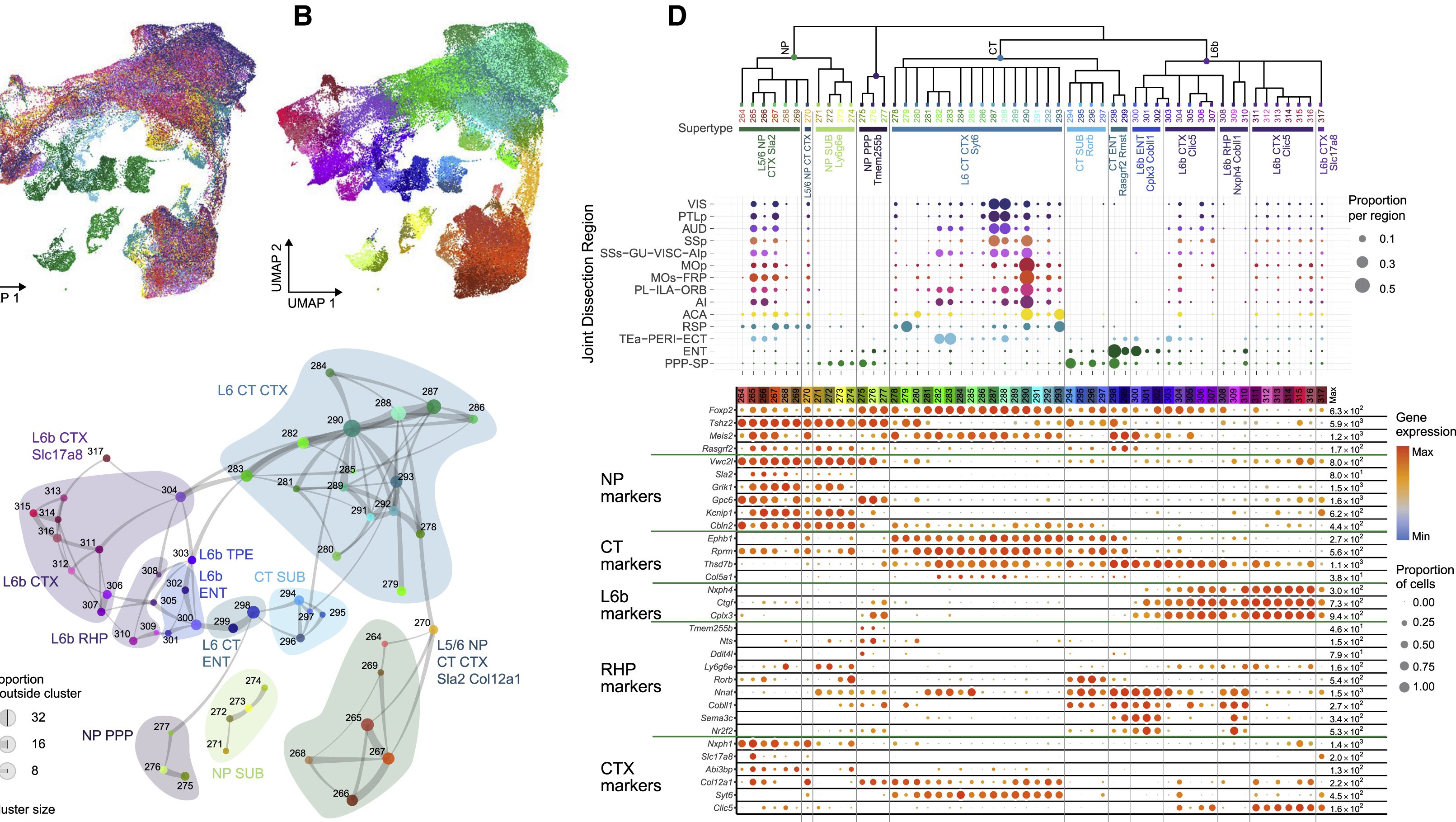
Pyramidal tract neurons (PT)
Note
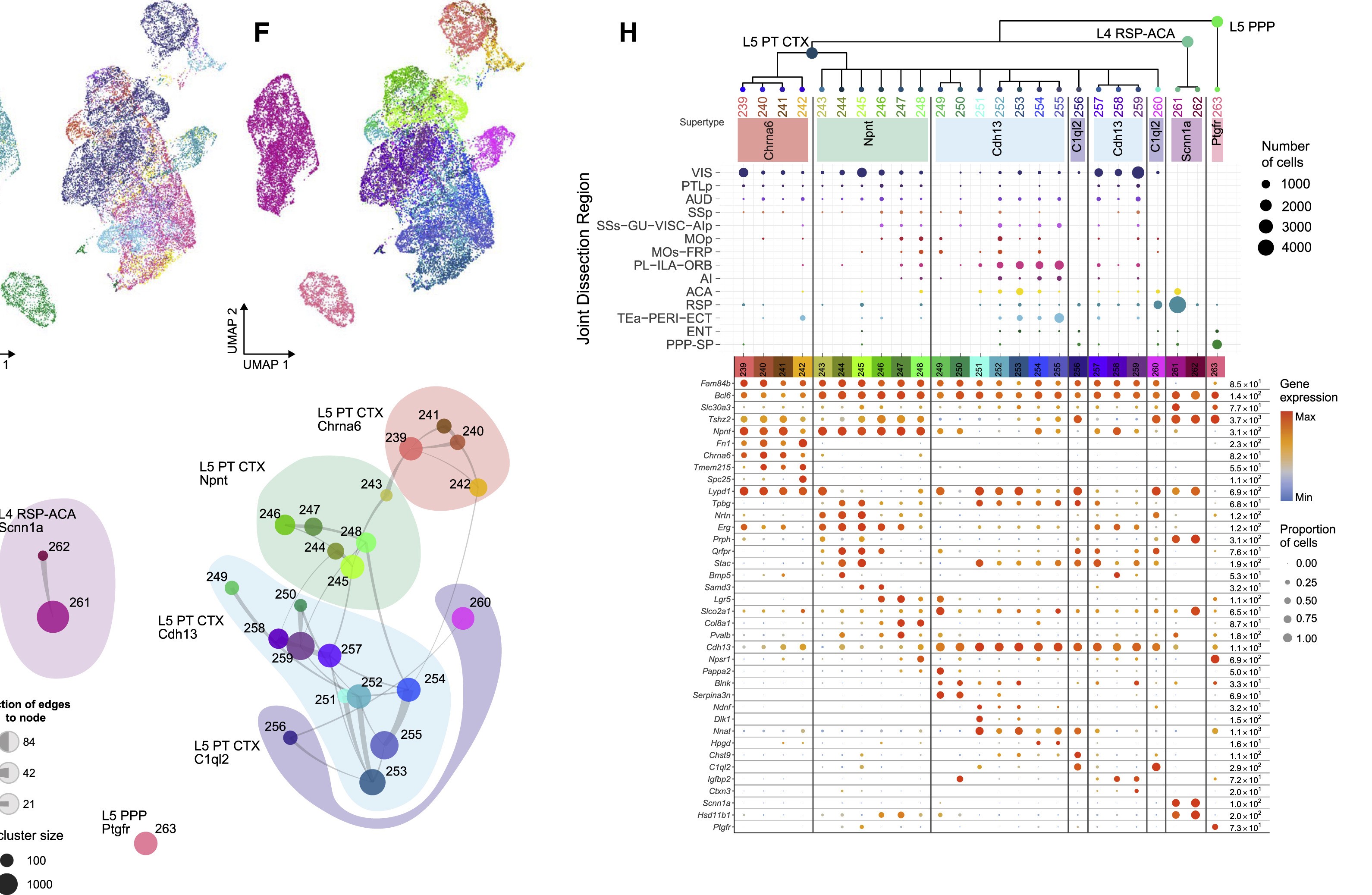
The Non-Neuronal class contains the Astro, Epend, Immun, Oligo, OPC, Vascu neighborhoods.
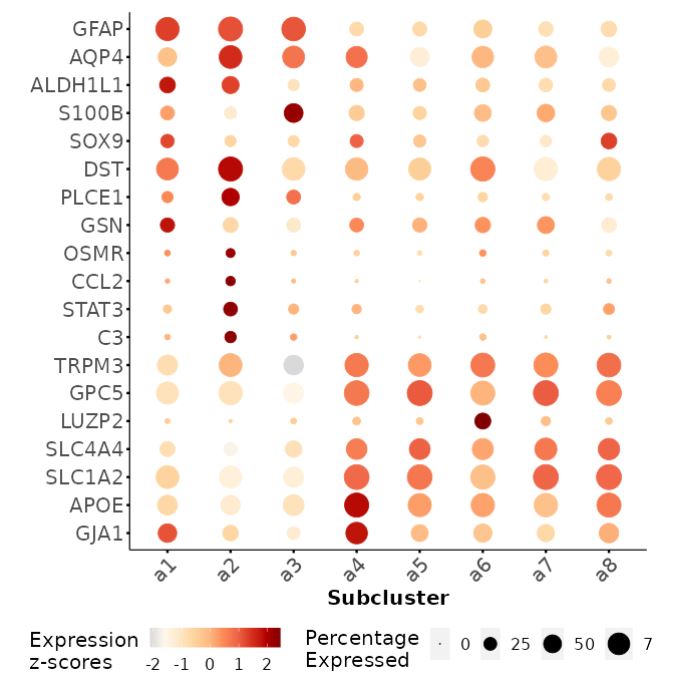
Astrocyte (Astro)
Ependymal cells (Epend)
TBD
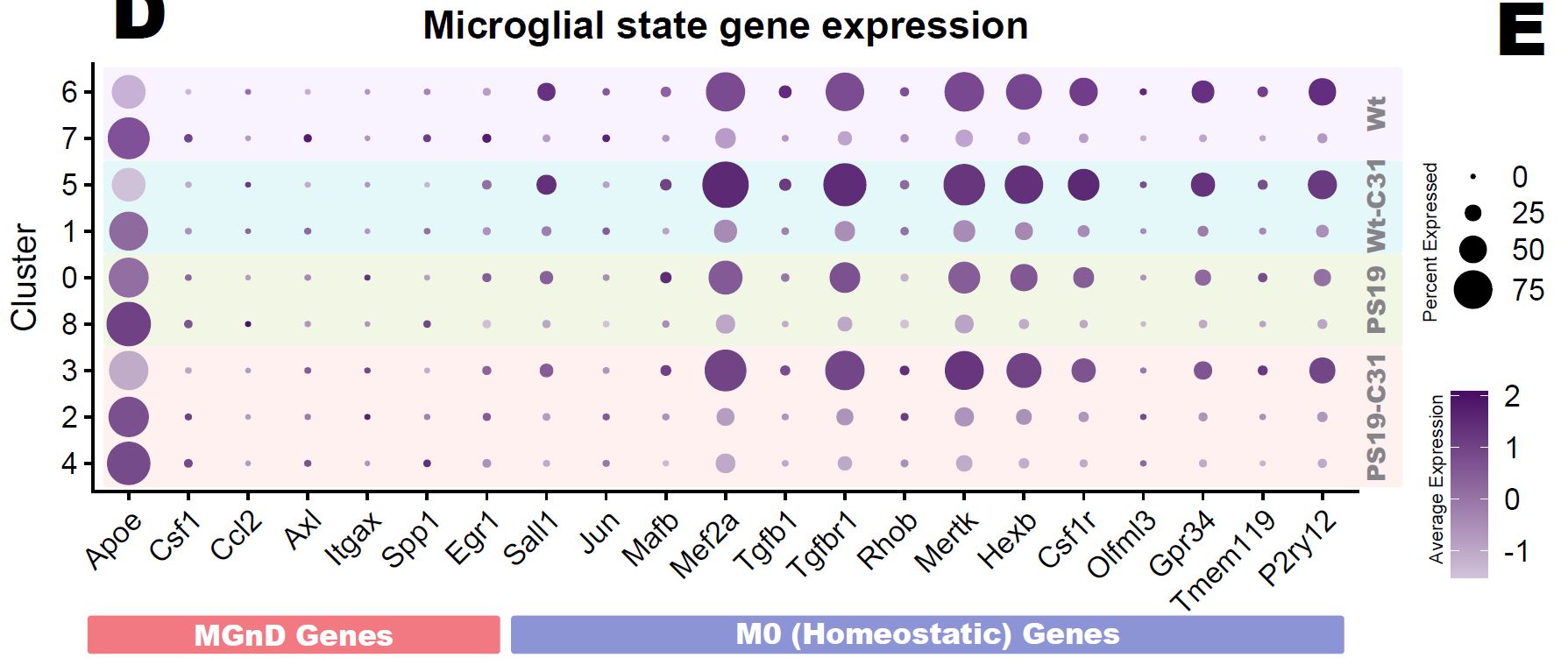
Immune cells (Immun)
Oligodendrocyte (Oligo)
Oligodendrocyte precursor cells (OPC)
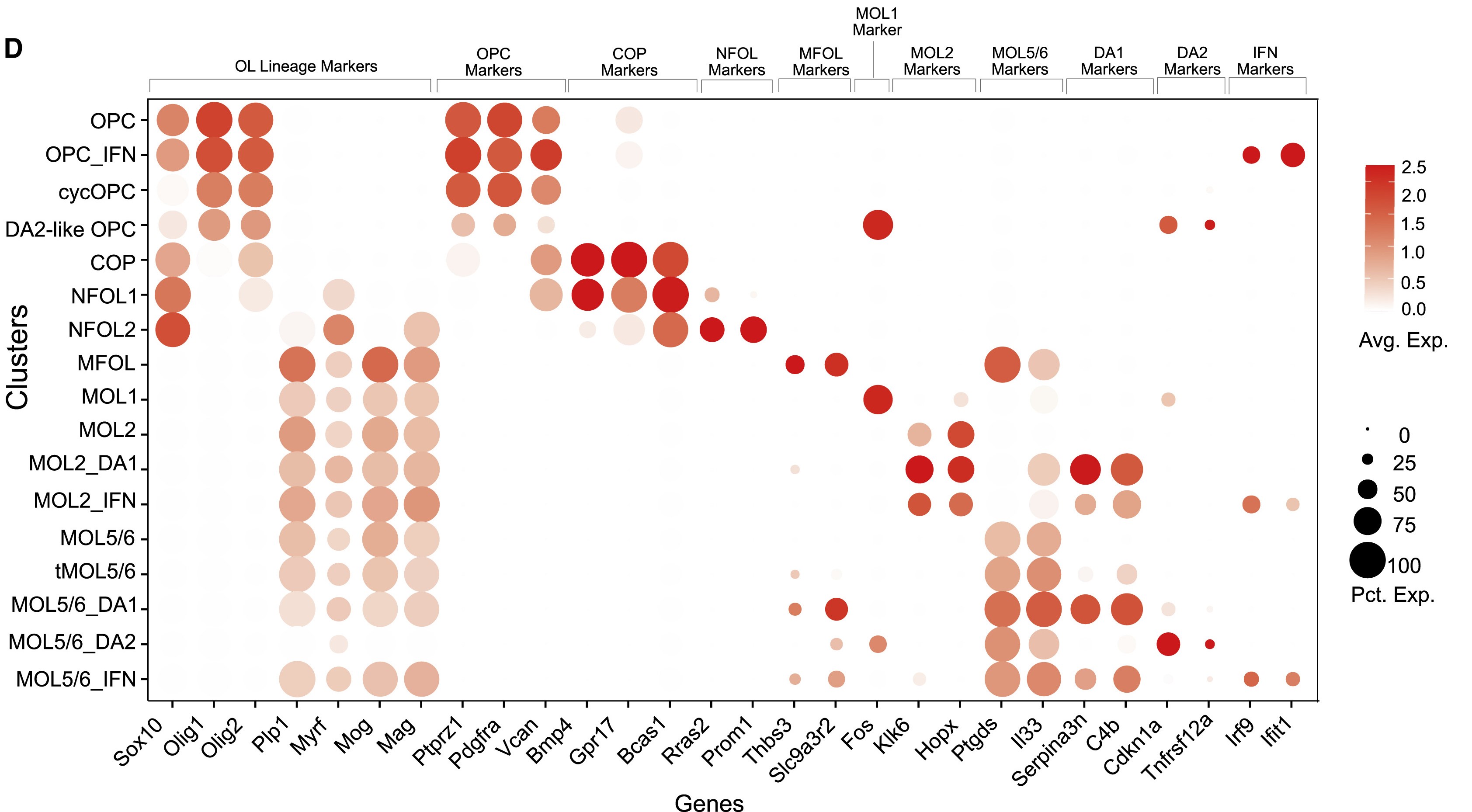
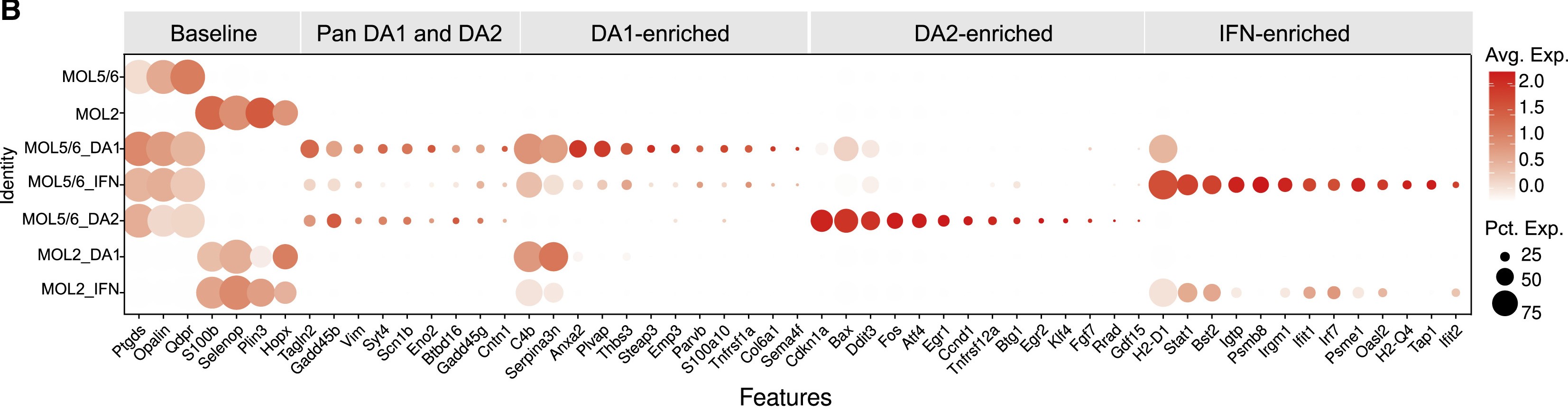
Vasculature cells (Vascu)
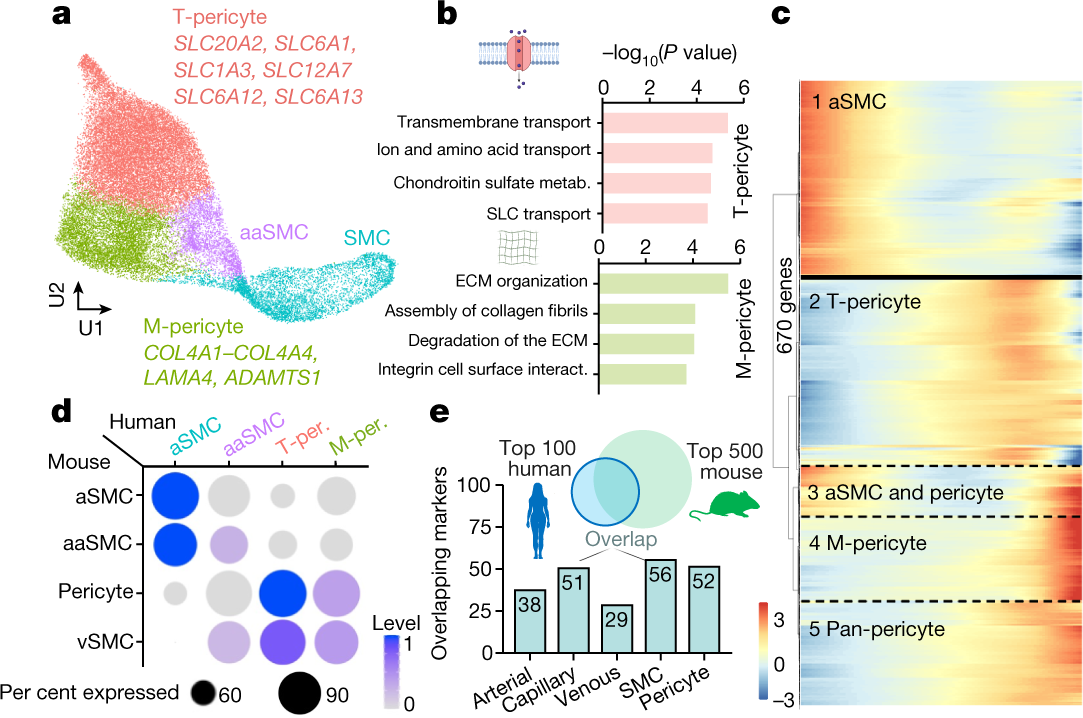
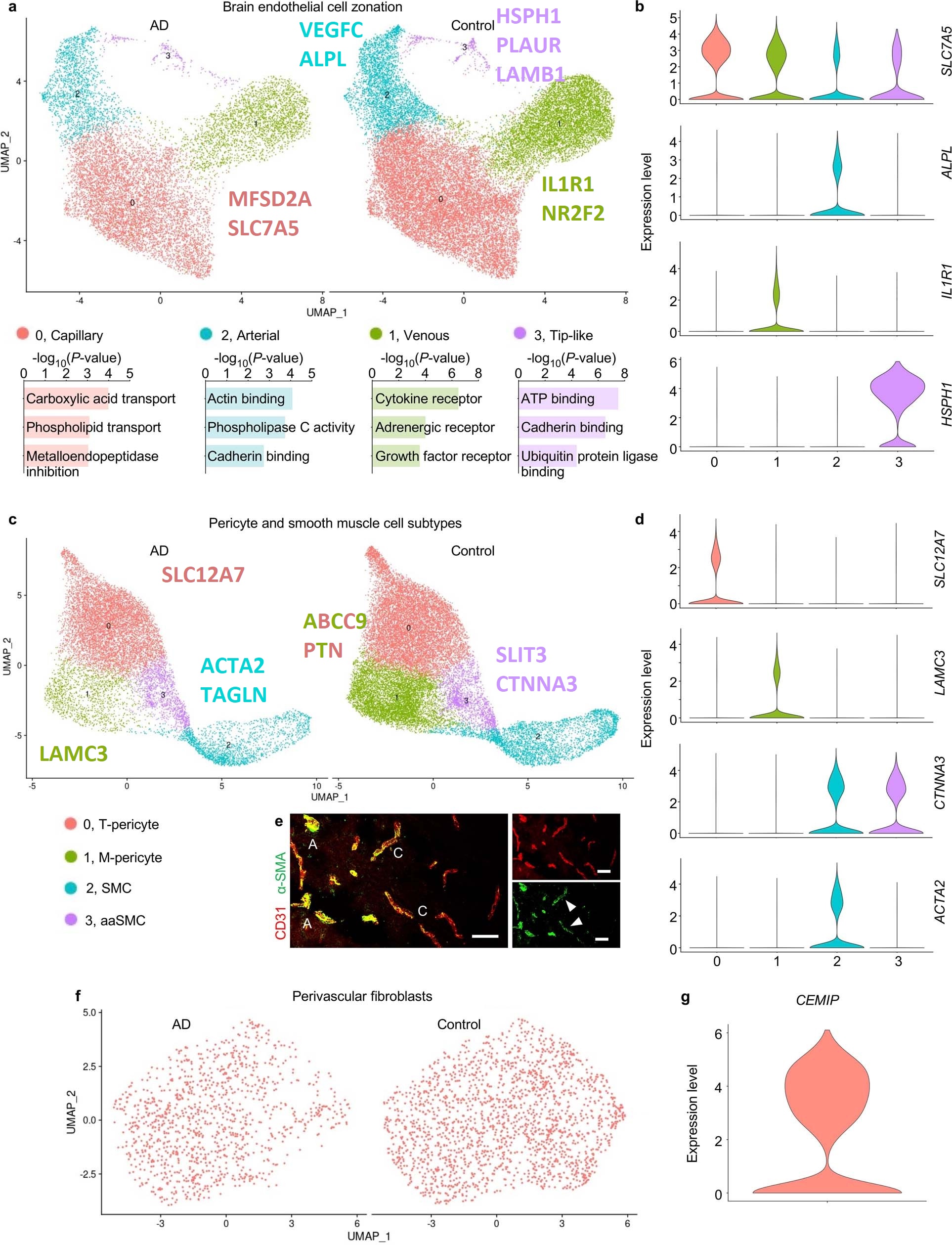
Gene set enrichment
In addition to looking at individual marker genes, we can also look at the set of top genes a cluster expresses and what terms are enriched from them to help shed some light on the identity of the cluster. gprofiler2 and Enrichr are two tools that take a list of genes as input and outputs enriched terms from multiple sources. Among others, gProfiler outputs Gene Ontology terms, biological pathways, regulatory motifs of transcription factors, and Enrichr outputs various pathways, ontologies, diseases/drugs, and cell types.
Low quality and dying cells
Something else to look out for are low quality or dying cells, which will be discarded after each round of annotation. Very low gene count and high mitochondrial fraction is a red flag for an artefact group.
Dying cells will also often have very few unique genes. In addition, they may be enriched for apoptosis, ubiquitination, and cell death, with some key markers being Ubb, Cmss1, Cst3, and Hspa8. You may see housekeeping genes Actb and Gadph, signifying that the cell type specific genes for these clusters are not very specific. They will also show a lack of good marker genes or markers for multiple sets. While these could be disease relevant (more cells dying in one group versus another), you also cannot entirely rule out they are dying cells from the experimental preparation/treatment, or are cellular detritus from lysing cells (SoupX tries to help with this), or other experimental artifacts. Thus, since their chimeric nature messes with clustering, we set them aside for our annotations, and will look back at experimental group membership later (in which case a proper cell death assay may need to be run).
Cluster consistency
Another thing to consider is the consistency of the cluster. We can visualize the cells of a cluster in the overall UMAP to determine its location in the bigger picture. This can help place the cluster in the context of the surrounding annotated cell types and give us a better idea of what the cluster is similar to.
Additionally, we can see how tightly or spread out the cells of the cluster is. If there is heavy spreading across the UMAP, the cluster can potentially be a chimeric cell type group, which will be discarded after each round of annotation. Chimeric clusters will have markers from multiple major groups, and the automated annotation will give inconsistent results.
Further subclustering
At the subclass level, if the cluster continues to harbor chimeric contamination, deeper subclustering may be needed to remove the bad cells.